¶ How to set up a unix account
To obtain a unix user name and password, create an account by registering at https://registrar.cs.huji.ac.il/account.
¶ Connecting to the hm server
Before connecting to the hm server, an SSH connection needs to be set up. Instructions for set-up can be found at Connecting from outside.
Every time you connect to the system, you will need to use a One-Time password (OTP) in addition to your unix (IDng) password. Instructions for obtaining OTPs can be found at Password and OTP.
It is helpful to set up an SFTP connection for easy file transfer between your desktop computer and the server. To do this with MobaXterm:
First, create a socks proxy on your computer, using MobaXterm's Tunneling option:

Clicking on “Tunneling" will open the below page, where you have to choose the “Dynamic port forwarding”.
Add a port for the SOCKS proxy on the Local clients (it's 8080 on the example, but you may use any port between 1024 and 65535). on the Remote server enter bava.cs.huji.ac.il as the remote host, and your user name (in this example: csuser):

Save the connection, Give it a name and start it on the next screen:

You would be asked for a password where you have to enter the OTP password.
Now that the SOCKS proxy is running, create a SFTP connection:
Click on “Session” and choose SFTP. Fill in the basic settings so that remote is the host you want to connect to (hm-gw for instance), username is your CS user. Click on “Advanced Sftp settings” and setup the “Proxy Settings” to “Socks5 Proxy”, Server: localhost, port: 8080 (in this example. In case you choose another port in the first step, fill in the same port here):
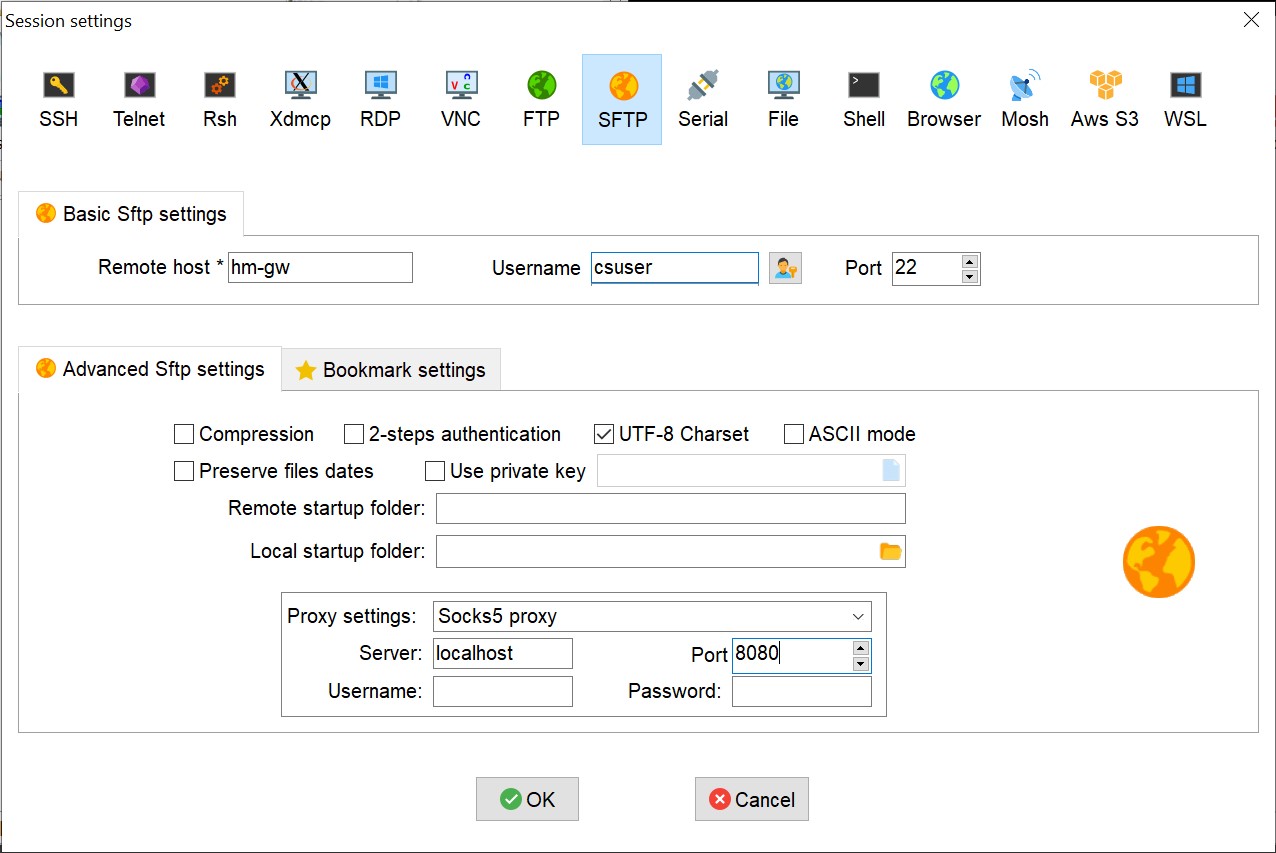
Click on OK. When starting the session you would be asked for your CS password (IDNG).
Before you start the transfer you will need to:
- Start the tunnel by clicking on 'Tunneling' and start the tunnel you created in the first step.
- Provide an OTP to open the tunnel
- Start the FTP connection by double clicking on the SFTP session
- Enter your CS password
The connection will eventually time out (you will see that the
button will no longer be pressed) after which you will need to re-do steps 1-4 to re-open it.
¶ Basic linux commands
For an introduction to some basic linux commands, take a look at the Intro presentation.
¶ Understanding the file system
The file system is divided into two main directories – /cs/icore and Lustre.
¶ /cs/icore
Your icore directory can be found in /cs/icore/<your unix user name>. Icore features snapshots so several previous versions of all files will be saved even after they are modified/deleted (assuming that the file system is up and running), however it has limited space.
- Icore should be used mainly to store your scripts.
- You should keep a copy of your scripts on Git, which allows you to save different versions. To use Git to back up scripts, see Github.
¶ Lustre
Your lustre directory can be found in /mnt/lustre/hms-01/fs01/<your unix user name>.
- Lustre should be used mainly to store the data that you are currently working on as it is meant for quick and efficient computations.
- IMPORTANT: lustre is not backed up!
This means that files on lustre that are accidentally deleted will be lost forever, so please be sure to back up all important files (e.g. on an external hard drive).
There are graphical interfaces that can make navigating the file system easier. For example, the nautilus file manager (can be opened with the command 'nautilus >& /dev/null &') and gedit text editor ('gedit >& /dev/null &'). For more information, take a look at the Intro presentation.
¶ Using slurm on the hm cluster
The hm cluster currently has 65 nodes on which jobs can run. Slurm is a job scheduler that decides which node(s) a job will run on based on the job's requested resources (e.g. number of cpus, amount of memory, etc…), the availability of idle nodes, and the priority of the user which depends on how many jobs were run in the past.
All jobs should be submitted as sbatch scripts to slurm where they will be queued and will start running as soon as all of their requested resources become available.
Some resource arguments that most users will want to tweak are:
| Argument | Default value | Maximum value | Description | Notes |
| -c, --cpus-per-task | 2 | 48 | number of cpus for job | Maximum depends on CPU used in on node, hm I-CORE has mostly nodes with 32 threads, but the newest nodes have 48 threads. |
| --mem | 50MB | ~64GB, ~128GB, ~256GB | memory allocation for job | Depends on memory of nodes, we have nodes with 64GB and 128GB of memory, you can't request all the memory since some is needed for the OS, if you want it all and don't care which node you can use: MaxMemPerNode. See below for more details. |
| -t, --time | 2 hours | 21 days | time limit for job | 21 days in long queue, 3 hours in short queue. |
| -N, --nodes | 1 | 65 | number of nodes to run job on |
For more information on all sbatch arguments, e.g., the proper format for writing time limits, etc…, read the sbatch manual https://slurm.schedmd.com/sbatch.html.
¶ Submitting a job to the slurm queue
There are 2 general ways to submit jobs using sbatch:
¶ One-liner
Resources can be requested as arguments on the command line, for example:
sbatch -N 2 --mem 8GB --wrap script.sh
The above command will submit script.sh as a job to slurm with two nodes and 8GB of RAM requested. The --wrap argument is needed so that the script (or command) is wrapped in an sbatch script.
To run a command with arguments, you need to enclose the command and its arguments in quotes, for example:
sbatch –o output.txt --wrap 'head -n 20 file.txt'
The above code will submit the command 'head -n 20 file.txt' as a job to slurm and will write the output of the command (the first 20 lines of file.txt) to the file output.txt.
¶ sbatch script (recommended)
Resources can be requested by adding special argument comments to bash scripts. These start with #SBATCH and then contain the sbatch argument, and will be parsed by sbatch until the script starts, so an #SBATCH argument written after the first command of the script will be ignored by sbatch. Here is an example of a bash script called my-sbatch-script.sh with sbatch arguments:
#!/bin/bash
#SBATCH --mem 10GB
echo "hello world"
# The following line will not be parsed by sbatch
#SBATCH --time 10
The above script can then be submitted to the slurm queue from the command line as follows:
sbatch my-sbatch-script.sh
my-sbatch-script.sh will be submitted as a job to the slurm queue with 10GB of RAM requested. The time limit for the job will be the default two hours since the time parameter will not be parsed by sbatch as it is written after a command in the script.
¶ Requesting resources
Make sure that your job does not request more resources than the maximum (please refer to table above). Jobs that request more than the maximum number of resources will not run.
Requests should be kept to as close to the needs of your job as possible for two main reasons:
- jobs are prioritized in the queue mainly based on requested resources, meaning that the more resources requested, the longer the job will take to start running since it needs to wait for all resources to become available
- while a job is running, all of its allocated resources will be unavailable to other users even if the job is not using them
¶ Requesting multiple nodes
Requesting multiple nodes for a specific job does not automatically divide the job among the nodes. There are a few ways to use multiple nodes simultaneously in order to save computation time:
- Using mpirun
- Using srun
- Using an outer submit script – this is recommended as it is the simplest method
¶ Using mpirun
If you know that you are running a program that depends on OpenMPI, you can use mpirun to run the program on multiple nodes in parallel – see the mpi manual (https://www.mpi-forum.org/docs/) for more information.
¶ Using srun
Srun can be used within an sbatch script to utilize a subset of the total requested resources. It can be used to run a command using multiple nodes in parallel which is most useful if you want to run a script/command on many files at the same time, each as its own job on one node, for example:
#!/bin/bash
#SBATCH -N 5
files=(file0 file1 file2 file3 file4)
for i in {0..4};
do
srun -N 1 –o output.$i.txt head –n 20 ${files[$i]} &
done
The above script will allocate five nodes as requested, and then will send five jobs to the slurm queue at the same time, each running on one node. The output of each job (the first twenty lines of the corresponding file) will be printed in the output.{i}.txt file. If you submit the above script as a job without using the srun command, for example:
#!/bin/bash
#SBATCH -N 5
files=(file0 file1 file2 file3 file4)
for i in {0..4};
do
head –n 20 ${files[$i]} &
done
Once the above job starts running, it will hold five nodes unavailable to other users as requested, but it will only run on one node.
¶ Using an outer submit script
The simplest way to run a script/command on many files at the same time, each as its own job on one node, is by using a for loop in a script that submits all of the jobs at once. For example:
#!/bin/bash
files=(file0 file1 file2 file3 file4)
for file in ${files[*]};
do
sbatch head.sh $file
done
The above script loops through all files in the list 'files' and submits the script head.sh (with 'file' as an argument) as an sbatch job. The head.sh script will contain the sbatch arguments needed to run on one file. For example:
#!/bin/bash
#SBATCH –o output.txt
#SBATCH –N 1
file=$1 #first argument from outer script
head –n 20 $file
The head.sh script will be run as a job on one node (based on the sbatch arguments written in the script) on each of the 5 files in parallel.
¶ Requesting multiple cpus
If you are running a program that supports multithreading, you can request multiple cpus (up to the maximum) and then run the program while specifying the number of cpus you want it to use as a parameter. For example:
#!/bin/bash #SBATCH –c 32
#request 32 cpus
#SBATCH –o output.txt
sample=$1
bowtie2 -p 32 -x $genome -U $sample -S $sam >& bowtie2.log
The above script can be submitted as a job that will run bowtie2 using 32 cpus on 'sample' (with the bowtie2 output saved in the file bowtie2.log).
¶ Requesting memory
If the amount of memory requested is not enough for the job, it will be killed with an error message that the job exceeded the memory limit and you will need to increase the memory request. There is currently no simple method to know how much memory a job will need before it runs. Once an sbatch job is running you can check its accounting information with the 'sacct' command.
¶ FAQs
¶ 1. How do I check the status of my job?
By running the command 'squeue –u username' with your username you will see all of your submitted jobs and their status (ST column). PD means your job is pending in the queue waiting to start, and R means it is running. For a list of all status codes, read the 'JOB STATE CODES' section of the squeue manual https://slurm.schedmd.com/squeue.html.
¶ 2. How do I cancel a job?
Jobs can be canceled by running the command 'scancel jobid' where the jobid is the number that slurm assigned your job when it was submitted which you can see by running 'squeue –u username' and looking at the JOBID column.
¶ 3. Why is my job taking so long to start running?
Jobs are prioritized in the slurm queue mainly based on the amount of resources requested. The more resources your job requests, the longer it will take to start running.
¶ 4. How much memory/cpus did my job actually use?
When a job is running, you can use the following commands to see its accounting information:
sacct – see the sacct manual for more details (https://slurm.schedmd.com/sacct.html).
sstat – see the sstat manual for more details (https://slurm.schedmd.com/sstat.html).
¶ 5. Can I submit a job and tell it to run at a later time?
You can tell a job what time you want it to start running with the parameter --begin:
#SBATCH --begin HH:MM MM/DD/YY
Additionally, you can add dependencies to your jobs so that, for example, job #2 will not start until job #1 satisfies the provided condition, by using the job id. This can be done in a bash script:
#!/bin/bash
jobid1=$(sbatch job1.sh)
sbatch –-dependency afterok:jobid1 job2.sh
The above script submits job1.sh as a job and saves jobid in the variable jobid1. It then submits job2.sh as a job only once job1 finishes running with an exit code of 0 as defined by the condition 'afterok'. For more information on possible conditions, etc… please refer to the sbatch manual https://slurm.schedmd.com/sbatch.html.
For more information, read the CS slurm wiki Slurm.